I
used three slides as shown below to illustrate the idea of informative DNAs in
my talk in last month’s workshop on genome and evolution in Naples, Italy.
The
antigenic sites in human influenza A virus mutate and turn over quickly, which
is critical for their survival or escape from human neutralizing antibodies and
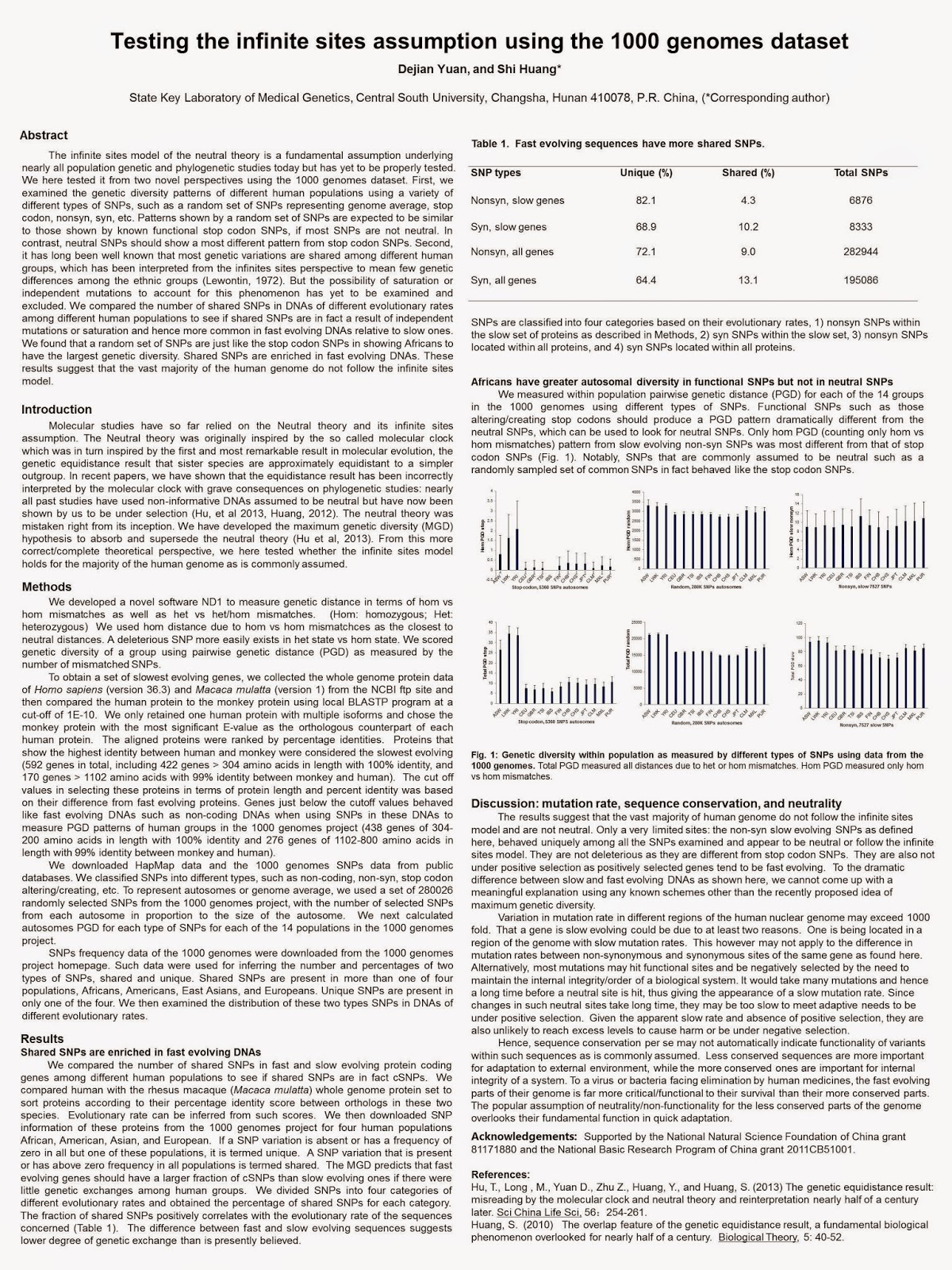
hence responsible for flu epidemics. As shown in Figure 1, two
amino acid positions in hemagglutinin (156 and 145, panel a and b) turned over
several times within a 30 year period, while two others (138 and 194, panel c
and d) stayed largely unchanged (Figure from Shih et al, 2007).
The
flu results illustrate two important points with regard to evolutionary
dynamics of a genome that have so far been grossly overlooked by the evolution and popgen field. First, fast evolving or less conserved DNAs are also functional
rather than neutral as they are essential for quick adaptive needs in response
to fast changing environments. Second, fast evolving DNAs turn over quickly and can be shown to violate the infinite sites model. Hence, they cannot be used for phylogenetic inference. If one uses the fast changing sites
in a flu virus to infer the phylogenetic relationship of the virus isolates responsible for different epidemics in a past period of say 10 years, one would reach the absurd conclusion that each epidemic was caused by a distinct type of flu virus with no genetic continuity among them rather than just
minor variations of the same type.
Mutation
rates in humans are of course much slower than that in a flu virus. But just
like a flu virus, there are also fast and slow changing sites (Figure 2). The time scales are different but the principle is the same. The fast
changing sites may turn over every few thousand years and in fact make up the
majority of the observed variant sites in humans when properly examined by us
(Figure 3). This is why the field of ancient DNA kept producing the absurd
pattern of no genetic continuity between people living in the same area but
from different periods of time. All of the published analyses have simply used
the wrong sites that are equivalent to the fast changing antigenic sites in a
flu virus. What one should be using are sites with very slow mutation rates,
like 1 mutation every 50,000 years. We have been busy reinterpreting the published
DNAs for several years now and hope to submit our work soon.

Figure
1. (a and b) Frequency changes at residue sites 156 (a)
and 145 (b) were highly dynamic. (c and d) Sites 138 (c)
and 194 (d) did not undergo major frequency change over time.
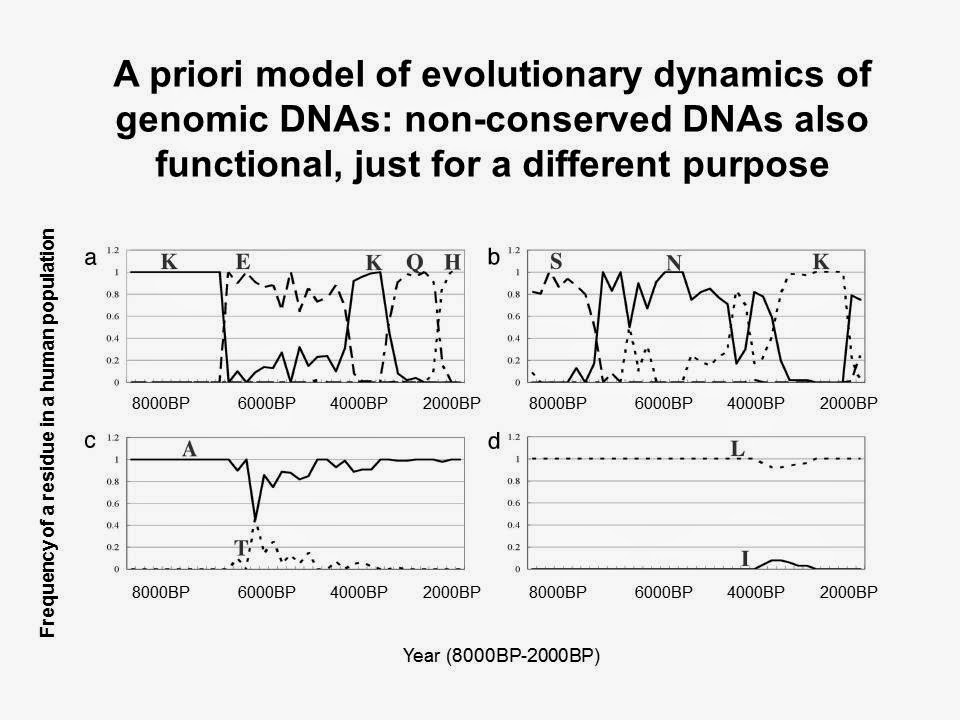
Figure 2. A priori model of evolutionary dynamics of human genomic DNAs.

Figure 3. Difference between slow and fast evolving sites. Shown are a piece of homologous DNA in three different individuals or species. In the fast evolving DNAs making up the vast majority of human genome, there is obvious and verifiable violation of the infinite sites model. These DNAs have abundant overlapped mutant sites where independent mutations have occurred on the same site in different individuals or species.
Ref.
Shih, C-C., Hsiao, T-C., Ho, M-S., and
Li, W-H. (2007) Simultaneous amino acid
substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc Natl Acad Sci U S A. 104:6283-6288.